Prepare candidate molecules
NOTE: this page is obsolete. See the About MolDB instead.
This topic describes how to prepare candidate molecules for virtual screening campaigns (and maybe other purposes). It describes how to select a subset of candidate molecules with suitable molecular properties that can be used as input for docking.
This topic shows how you can perform these tasks with the Squonk Data Manager user interface. You can also run these as command line tasks as described here.
Prior tasks
Before running this you will need to have performed these use cases:
- Add libraries to the sharded file system (note: this has probably already been done for you).
After tasks
After running you might be interested in running these use cases:
- Use case Docking with rDock
Background
The sharded file system contains millions of purchasable or synthesisable molecules that you might want to use in a virtual screening campaign. This is the haystack in which you are trying to find the needle. Searching the entire haystack is not practical. Instead you want to create a subset that contains the molecules you might be interested in finding. This use case describes how you can do this. Typically you might want to restrict your candidate molecules:
- to have a limited range of heavy atom counts, as you probably already have an idea of what size of molecules might fit your active site
- to contain rings, or certain numbers or types of rings
- to not have too many (undefined) chiral centres
- to be not too planar
- etc. etc.
This use case lets you specify these criteria, to optionally select a diverse subset of these to reduce the number of molecules whilst maximising your chances of finding something, and to enumerate microstates, tautomers and stereoisomers of those molecules, generating a suitable starting conformations for use in docking.
The filter job lets you select the suitable subset.
The max-min-picker job lets you optionally select a diverse subset of those molecules
The prep-enum-conf-lists job lets you prepare lists of molecules that have not yet been enumerated
The enumerate-candidates job lets you enumerate the microstates, tautomers and stereoisomers of those molecules, generating a suitable starting 3D conformation.
The generate-low-energy-conformers job lets you generate low energy conformers of each enumerated molecule.
The assemble-conformers job lets you generate a SD file ready for further work (e.g. as input for docking).
This use case describes how to combine these steps and create a useful set of candidate molecules for further processing.
Workflow
Step 1: Filter job
Here you use the filter job to specify the selection criteria for the molecules you want to screen. For the inputs select the suitable directory or directories inside the molecules directory (or wherever the sharded filesystem was placed). Also specify a name (which can be a path with subdirectories) for the output file (filtered.smi is the default).
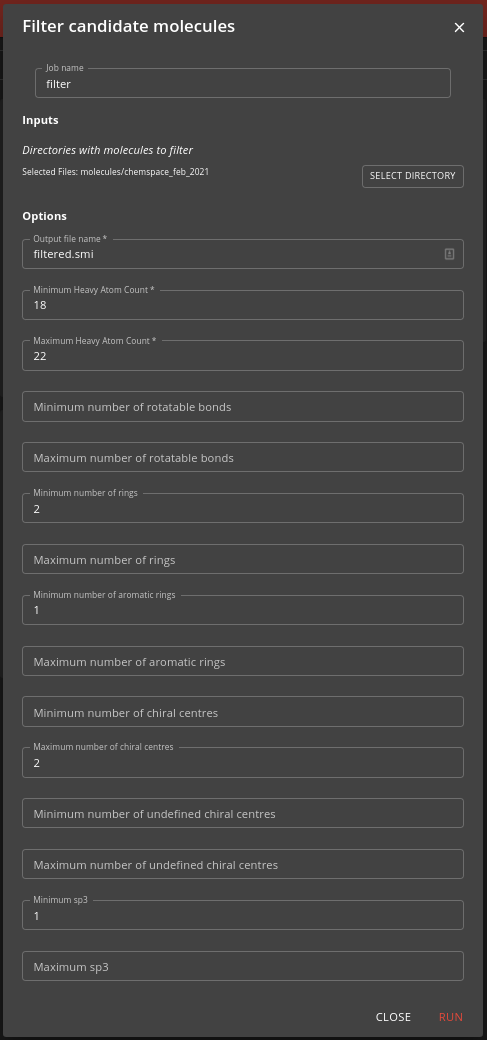
The output is a files containing the SMILES, the UUID and the SHA256 digest for the selected molecules. The job may take a few minutes to complete. You can view the progress by looking at the job’s task.
output.
Step 2: Diverse subset selection
If your filtered set of molecules is too big then you can use the max-min-picker job to select a diverse subset of molecules with the number of molecules that you want. The MaxMin Picker picks the first molecule at random, and then successively picks the molecule that is most dissimilar to those already picked.
Specify your filtered molecules (the output from the previous step) as the input and the number of molecules you want to be picked. You can also optionally specify a set of molecules that are already picked, and to stop picking once the similarity threshold rises above a certain level. Also specify a name (which can be a path with subdirectories) for the output file (diverse.smi is the default).
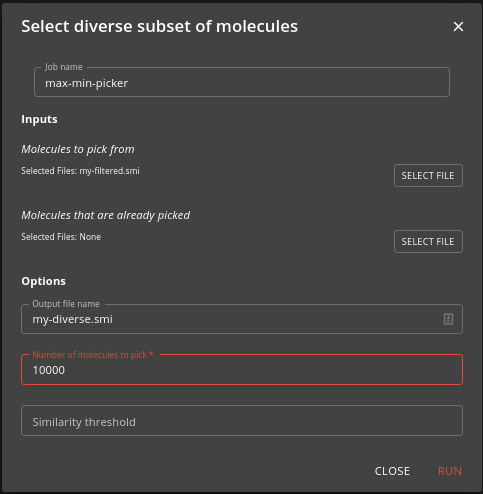
The job may take a few minutes to complete.
Step 3: Prepare list of molecules needing enumeration and low energy conformer generation
This step used the pre-enum-conf-lists job to prepare lists of molecules that have not yet been enumerated or have low energy conformers generated. These are stored in the sharded file system so that they only have to be generated once, as the process is computationally intensive (especially for the low energy conformer generation). For the list of candidate molecules you prepared in the last two steps some might have already been processed, others not. This step identifies those that need to be processed.
Specify the input molecules and names for the two output files.
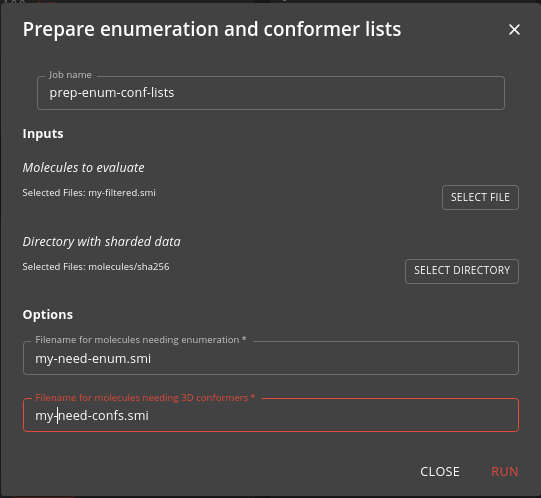
The job is relatively fast unless you are processing a very large number of molecules.
Step 4: Enumerate molecules
This step takes the list of molecules that need enumerating from the previous step and uses the enumerate-candidates job to enumerate the tautomers, microstates and undefined chiral centres. It is computationally demanding and uses a Nextflow workflow to parallelise the processes across the Kubernetes cluster.
Choose the need-enum.smi file that was created in the previous step as the input.
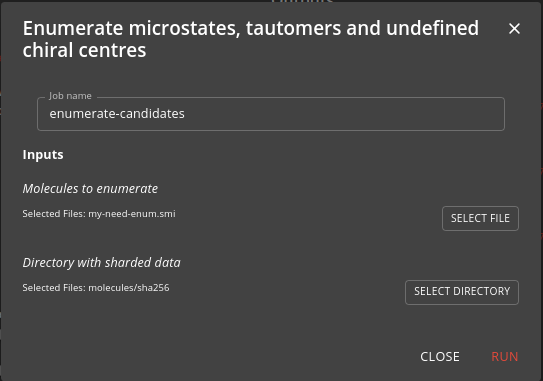
All output is written to the sharded file system.
Step 5: Generate low energy conformers
This step uses the generate-low-energy-conformers job to take the enumerated molecules from the previous step and generate low energy conformers. Some downstream processes, such as docking do not need this step to be performed. It is computationally very demanding and you should filter your molecules as best you can if needing to perform this step. It uses a Nextflow workflow to parallelise the processes across the Kubernetes cluster.
Choose the need-confs.smi file that was created in step 3 as the input. Step 4 must already have been run.
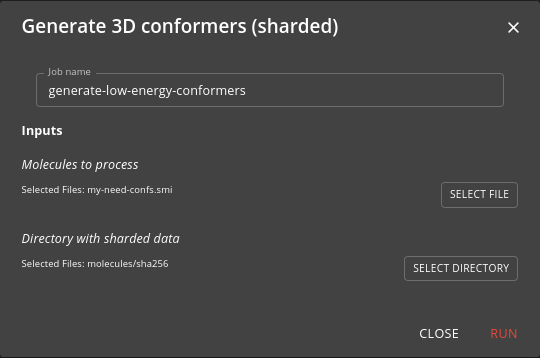
All output is written to the sharded file system.
Step 6: Assemble conformers
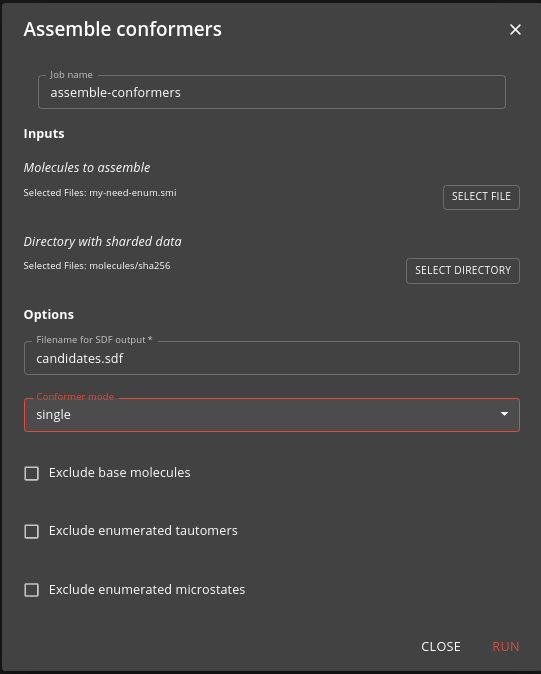
Typically you need to assemble the outputs or steps 4 and 5 into a SD file that will be input to a downstream job such as docking. The assemble-conformers job does this for you.
As input you specify the need-enum.smi or need-confs.smi files output from step 3. The job operates in one of two modes:
- single - assembles the single conformer for the molecules enumerated in step 4. This, for instance, can be used as input for docking.
- low-energy - assembles the multiple low energy conformers for each molecule generated in step 5. This, for instance, can be used as input for ligand based virtual screening.
Tun the job in whichever mode you want, or run it twice in each mode if you need both types generated.
In addition to specifying the mode, you can also specify whether to exclude the base molecule, the enumerated tautomers or the enumerated microstates.
The resulting SD files contain fields with useful information about how the conformer was generated, and its relationship to the molecules in the sharded file system.