rDock docking use case
This topic describes how to run jobs to perform molecular docking using the run-rdock job.
The same approach can also be used for other docking tools e.g. smina .
Prior tasks
Before running this you will need to have performed these use cases:
- Add libraries to the sharded file system using the shard job (note: this has probably already been done for you).
- Use case: Prepare candidate molecules to create the candidate molecules you want to dock.
The result will be a SD file containing a single conformer of the enumerated molecules you choose for screening. For instance, you might have selected molecules withing a particular heavy atom count range and with a certain number or rings etc. You might have selected a diverse subset of from these. You will have enumerated the tautomers, microstates and undefined chiral centres of these molecules, resulting in a single conformer of each. Those conformers are the input to docking.
Docking
As well as the SD file containing the candidate molecules, the The rDock tool needs the receptor in mol2 format, and active site definition file (the .as file) and a configuration file (the .prm file) that links these together and defines the docking parameters. Precise details can be found in the rDock documentation, but you can use the
prepare-rdock job job to create a basic configuration. This job does the following:
- generates the .as file with the active site definition, by using an exemplar ligand to define the bounds
- creates the .prm file with the necessary configuration
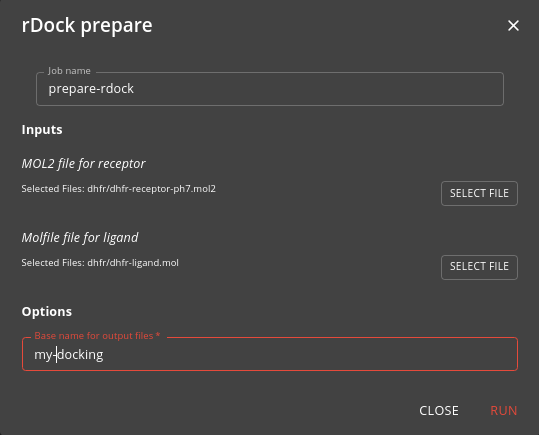
Running this job will run the rbcavity program to generate the .as file and will create the .prm file.
The base name is what you specify (default docking). The typical docking.prm file that you generate will look like this:
RBT_PARAMETER_FILE_V1.00
TITLE rDockdocking
RECEPTOR_FILE /data/dhfr/dhfr-receptor-ph7.mol2
RECEPTOR_FLEX 3.0
##################################################################
### CAVITY DEFINITION: REFERENCE LIGAND METHOD
##################################################################
SECTION MAPPER
SITE_MAPPER RbtLigandSiteMapper
REF_MOL /data/dhfr/dhfr-ligand.mol
RADIUS 3.0
SMALL_SPHERE 1.0
MIN_VOLUME 100
MAX_CAVITIES 1
VOL_INCR 0.0
GRIDSTEP 0.5
END_SECTION
#################################
#CAVITY RESTRAINT PENALTY
#################################
SECTION CAVITY
SCORING_FUNCTION RbtCavityGridSF
WEIGHT 1.0
END_SECTION
That may suffice for basic docking, but you should consider what is needed based on the rDock documentation. You can either edit this file or create a new one according to your needs.
Once you have configuration you need you can use the run-rdock job to run the docking, specifying the SD file you generated earlier as input as well as other docking parameters.
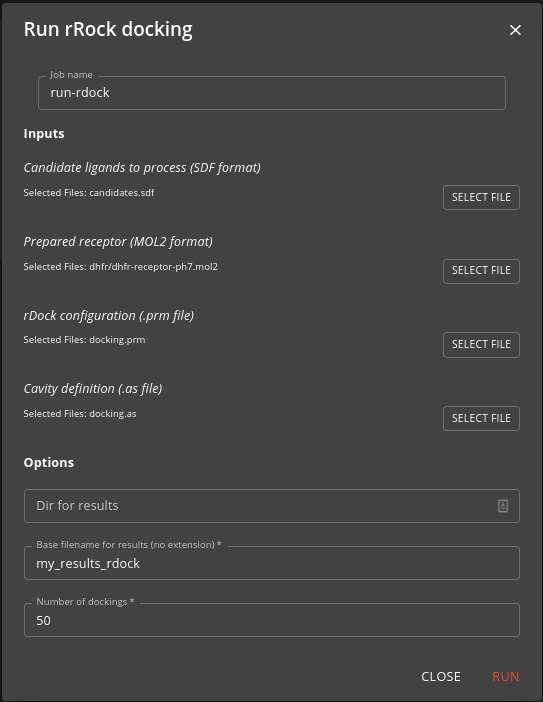
The job is run using Nextflow to parallelise the docking across the cluster. It will take some time if you have many candidate molecules to dock. The result will be a SD file containing the docked poses (as many poses for each molecule as you specified in the job options).
Post execution options
- Use the oddt-score-interactions job to investigate the interactions each pose makes with the protein
- Use the filter-sdf job to reduce the contents of the output SD files to the best scoring record within a group (e.g. the best of the n generated poses)