This post will describe how Squonk jobs can be used to prepare molecules for virtual screening.
The basic flow will prepare a single conformer of the candidate molecules, ready for docking, but variations on this flow will also be discussed. In another post we will describe how to use rDock to dock these molecules. rDock requires a starting 3D conformation of each candidate molecule, which is what our basic flow generates.
The input molecules come from ChemSpace and are as SMILES. They look like this:
O=C(CCc1c[nH]c2ccccc12)OCc3ccccc3 CSMB00000000002
O=c1n(Cc2ccccc2)c(=O)c3n(Cc4ccccc4)cnc3n1Cc5ccccc5 CSMB00000000003
COCN1C(=O)NC(C)(C)C1=O CSMB00000000008
O=C(CCN1C(=O)c2ccccc2C1=O)OCc3ccccc3 CSMB00000000014
N#Cc1c(NCc2ccccc2)sc3CCCCc13 CSMB00000000015
C=CCSc1nnc(NCc2ccccc2)s1 CSMB00000000017
O=c1oc2ccccc2n1Cc3ccccc3 CSMB00000000021
CC(NC(=O)c1ccccc1)C(=O)OCc2ccccc2 CSMB00000000023
CCCCNc1nnc(SC)s1 CSMB00000000029
CCCCNc1nnc(SCC)s1 CSMB00000000030
Very simple, a SMILES and an identifier separated by a tab.
We start with 100,000 of these, but we make the assumption that we only want to dock a small subset of these to limit the computational need. Our flow is like this:
- filter the inputs, removing duplicates and keeping only molecules of the size we want
- create a small diverse subset
- enumerate potential microstates, tautomers and chiral centres
We’ll also describe a potnential subsequent step to generate low energy confomers, but these are not needed for docking.
Step 1: filter the inputs
We do this with the rdkit-dedup job. The primary purpose of this job is to de-duplicate a set of molecules, allowing the molecules to be stripped of salts and other small components. The biggest fragment is identifed, and you can select whether to do this using the heavy atom count (HAC) or molecular weight (MW). We use HAC.
Find the job in the Apps/Jobs tab by typing dedup into the search box adn then click on the RUN button. Set the options like this:
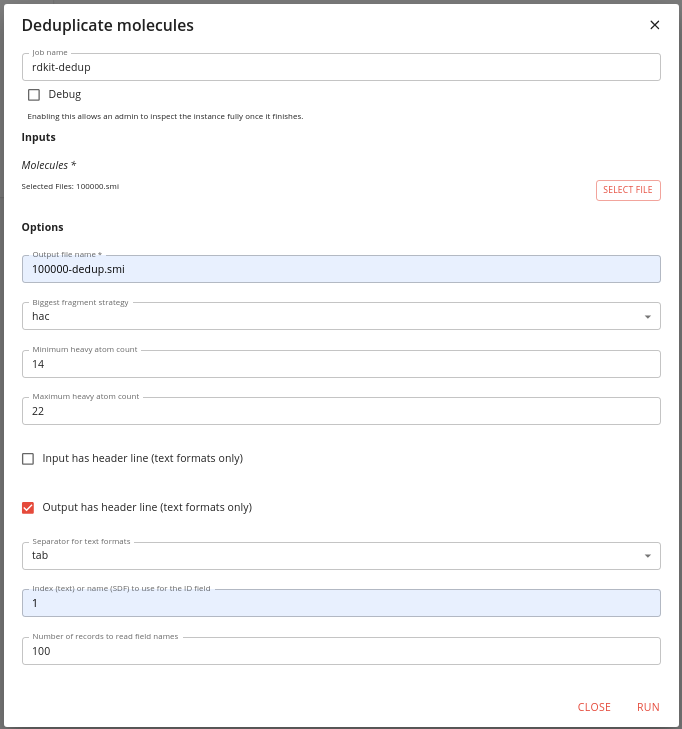
You need to specify the input file and a name for the output file.
We specify that we want to use HAC to select the biggest fragment. We want to only include molecules within a particular size range. Here we choose a minimum of 16 heavy atoms and a maximum of 22. We specify that the input files does not have a header (or you will loose the first molecule), that you want the output file to have a header. Also we specify that the files are tab separated, and the ID column is number 1 (this is zero indexed so the SMILES is column 0 and the ID column 1).
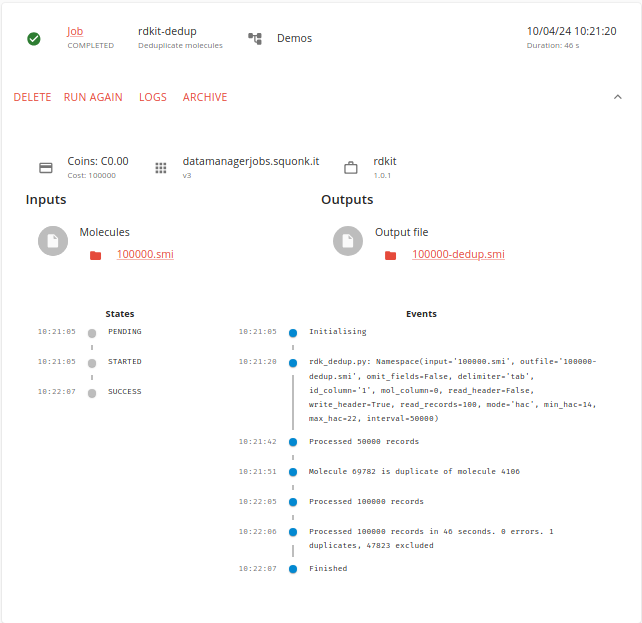
The job runs quite quickly and we are told that 47823 molecules are excluded and there was 1 duplicate.
We can view the output in the text viewer if we want.
Step 2: generate a diverse subset
For this we use the max-min-picker job, which is also based on RDKit.
Type maxmin into the search box to find this job and click the RUN button.
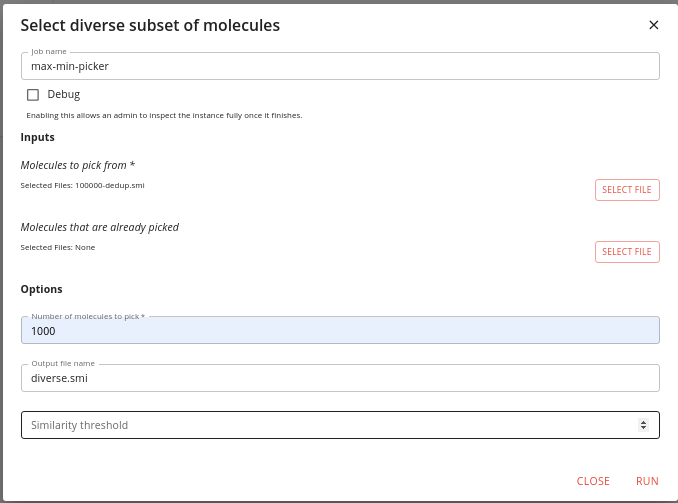
You need to specify the output of the previous job as the input to this one, and a name for the output.
Do not specify anything for the Molecules that are already picked input.
Specify how many molecules to pick (we choose 1000) and then click RUN.
Again, this job runs quite quickly as we are not picking many molecules.
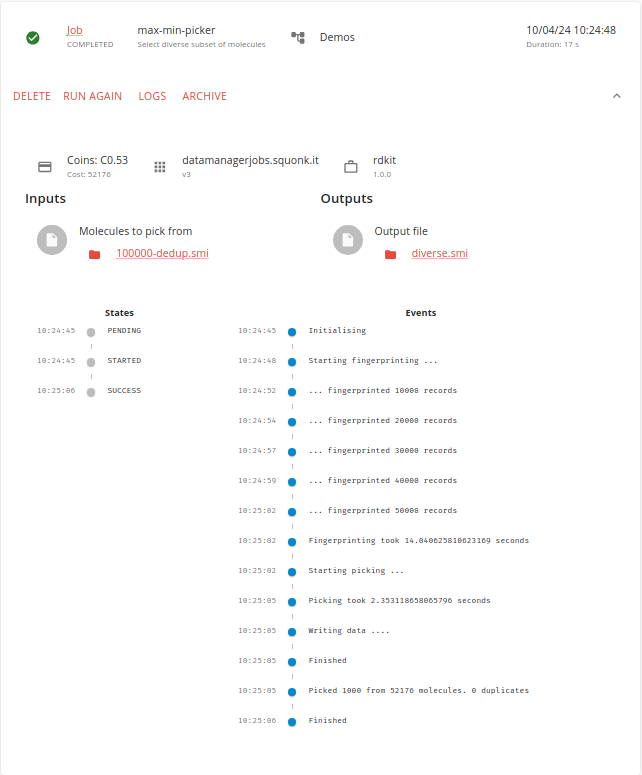
We now have 1000 diverse molecules selected from the 100,000 original ones.
Again, we can view the outputs with the text viewer if we want.
Step 3: enumerate molecular forms.
This step allows to enumerate different forms of these molecules. This includes microstates (charge forms), tautomers and chiral centres that are undefined. If you are concerned about the computational resource needed you might not want to enumerate all these, but clearly stereochemistry, charge and tautomerisation can have a big impact on the binding of a ligand.
The job is called enumerate-candidates and can be found by typing enumerate-c into the search box. Make sure you select the right job.
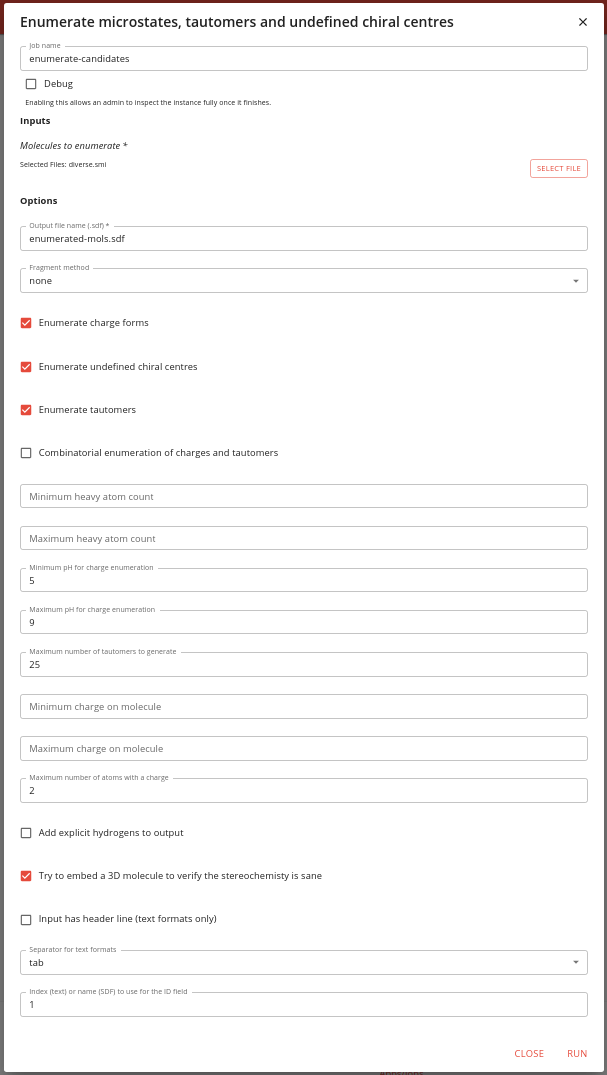
The options for this job are quite complex. Specify thee input as the output of the previous job, and a name for the output file.
There is the option to select how to define the biggest fragment, but we have already done this in the rdkit-dedup job, so we can set this to none.
Similarly, we do not need to specify values for the min and max heavy atom count as we’ve done this earlier.
Select whether you want charge forms, undefined chiral centres and tautomers to be enumerated. You can also specify the combinatorial enumeration of charges and tautomers, but this can lead to generation of very many molecules.
You must specify the min and max pH values for charge enumeration. We leave these as the default values of 5 and 9.
We leave the maximum number of tautomers to generate for each molecule to 25. This prevents generating and insane number of tautomers.
There are also options for controlling the generation of charge forms. You can specify a min and max charge for the molecule, and the max number of atoms with charges. We just choose a max number or charges of 2.
You can specify whether to add explicit hydrogens to the output molecules (we say no).
It is generally sensible to try to embed a 3D molecule, firstly because this gives you a sensible 3D structure, but also as it allows to filter out molecules with insane stereochemistry (often seem with caged structures).
Finally there are options for handling text files, whether there is a header line (there isn’t), what the separator is (tab) and which column is the ID (column 1, zero indexed).
With all that set we can run the job.
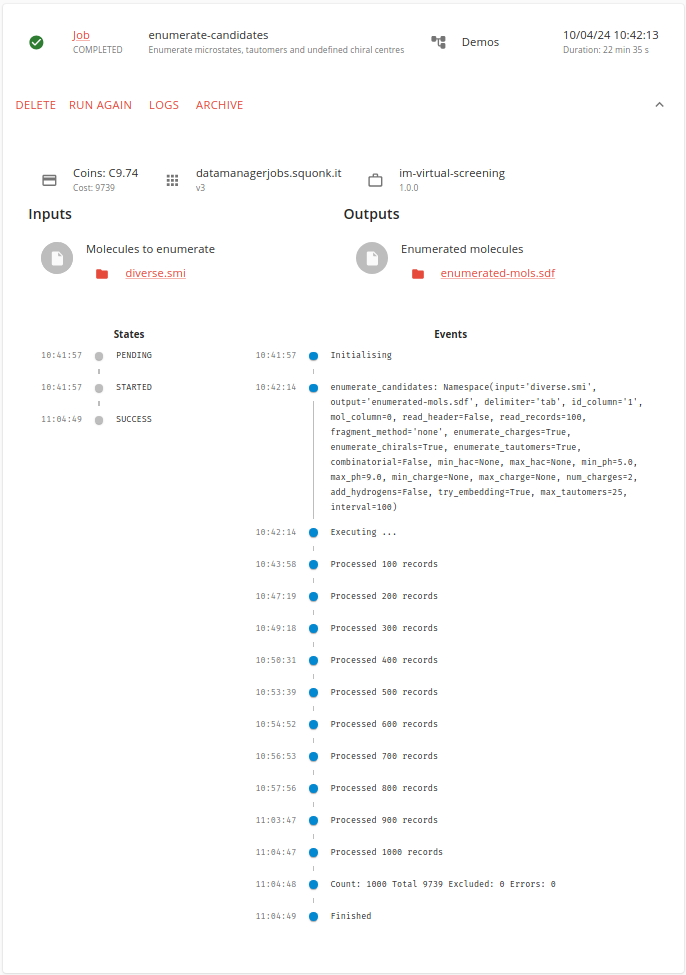
This does take a little while to run, but eventually we find that we have enumerated 9,739 molecules.
These can be used as the input to our docking.
Step 4: conformer generation
We don’t need to generate multiple confomers for docking, but for other approach this is needed. For instance in doing rigid ligand alignment (ligand based virtual screening). So we will briefly describe this here.
We choose to use the enumerated molecules as input, but we could have used non-enumerated molecules instead.
The job is called generate-low-energy-conformers. It uses RDKit’s ETKDG conformer generator.
Some of it’s options are the same as the earlier jobs. The ones specific to conformer generation are the maximum number of conformers to generate, the number of energy minimisation steps and the RMS threshold for excluding similar conformers. We use defaults, except that we limit the number of conformers to 25 just so that we can run this job more quickly. If this value is not specified the rules defined by Inhibox are use, which are to generate up to 50 conformers for molecules with 7 or less rotatable bonds, up to 200 for molecules with 8 to 12 rotatable bonds and up to 300 for molecules with more than 12 rotatable bonds.
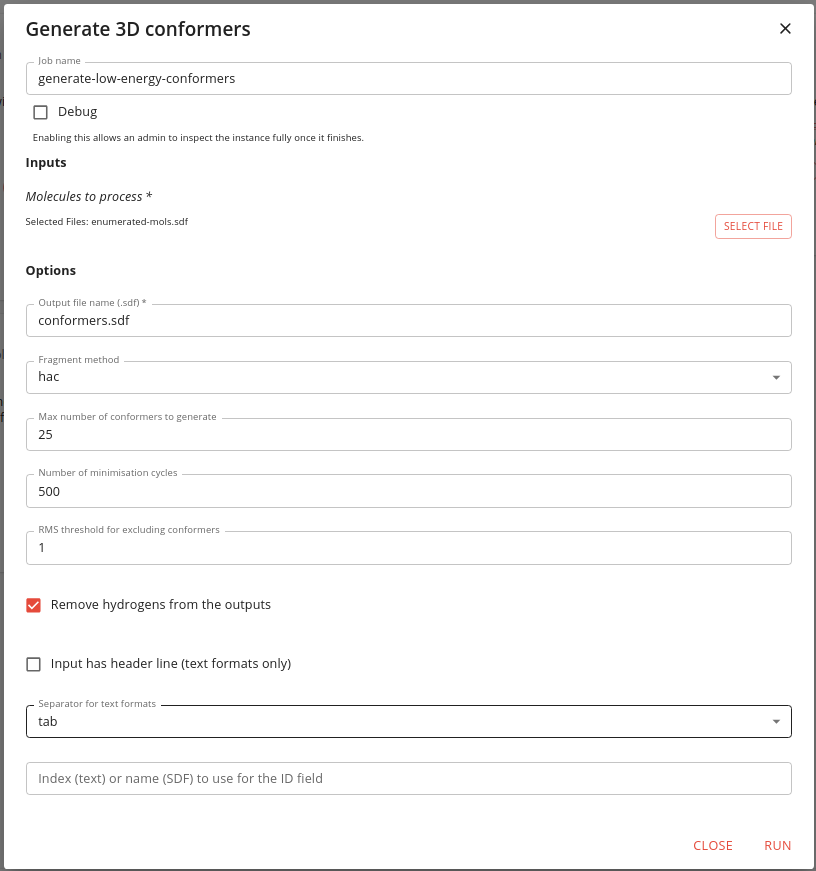
The job takes some time to run. The initial execution looks like this:

It finishes like this:

Parallel execution
We notice that the enumerate-candidates and generate-low-energy-conformers jobs take some time to run. This is because they are computationally demanding and run as a single process, each molecule at a time. To address this we also have parallelised versions of these jobs, which use Nextflow to manage the parallel executionn of the Kubernetes cluster in which all this is running.
The jobs are called enumerate-candidates-multi and generate-low-energy-conformers-multi and look almost the same as their simple counterparts. The only extra option is to specify the Size of chunks to split. This controls how the splitting of the inputs is done. With the default of 1000 then the inputs are split into separate files of 1000 molecules, and then each file is run in parallel, with all the results being combined back into a single output file. Depending of the size of the Kubernetes cluster in which this is running, this can provide a big improvement in execution time.
Flexible operation
The enumerate-candidates and generate-low-energy-conformers are the key jobs described here. They are described in the context of the flow that we illustrate here, but the can be run independently. Each one can take a SMILES text file of a SD-file as input, and they do not need to be run as described here.
MolDB as an alternative
The flow described here is really best suited for standalone runs where the number of molecules is kept to a relatively small number. It is obviously not efficient for every user to enumerate the same molecules as this is computationally demanding/ This is why we have created MolDB, a centralised database of molecules, their enumerated forms and low energy conformers. Whilst slightly less flexible than the approach described here, it might be more suitable if you want to avid doing all the heavy lifting yourself.
You can find out about MolDB here.